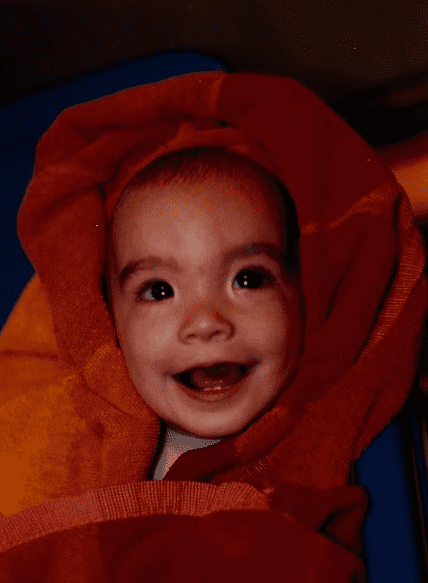
Caratteristiche
La sindrome cri-du-chat, detta anche monosomia 5p o monosomia parziale, è una malattia genetica rara, dovuta alla delezione del cromosoma 5. Il nome si riferisce al caratteristico pianto, che ricorda quello felino, dei bambini affetti. E’ contraddistinta, spesso, da caratteristiche facciali distintive, sviluppo ritardato e disabilità intellettiva. Fu descritta, per la prima volta, da Jérôme Lejeune, un medico francese, nel 1963.
Eziogenesi e patogenesi
La sindrome di cri-du-chat è stata descritta per la prima volta nel 1963 dal Dr. Jerôme Lejeune in tre pazienti non imparentati. La sindrome è causata dalla delezione 5p causata dalla perdita dell’estremità del braccio corto del cromosoma 5. Nella maggior parte dei casi la delezione è sporadica o casule, quindi l’insorgenza avviene in modo spontaneo. Nella restante parte la delezione è dovuta alla segregazione ineguale della traslocazione bilanciata. Si ritiene che due geni in questa regione, la Semaphorine F (SEMAF5A) e delta-catenina (CTNND2), siano potenzialmente coinvolte nello sviluppo cerebrale. La loro delezione può essere associata al ritardo mentale dei pazienti affetti dalla sindrome di cri-du-chat. La delezione del gene della trascrittasi delle telomerasi (hTERT) può contribuire anche ai cambiamenti fenotipici nella sindrome del cri-du-chat.
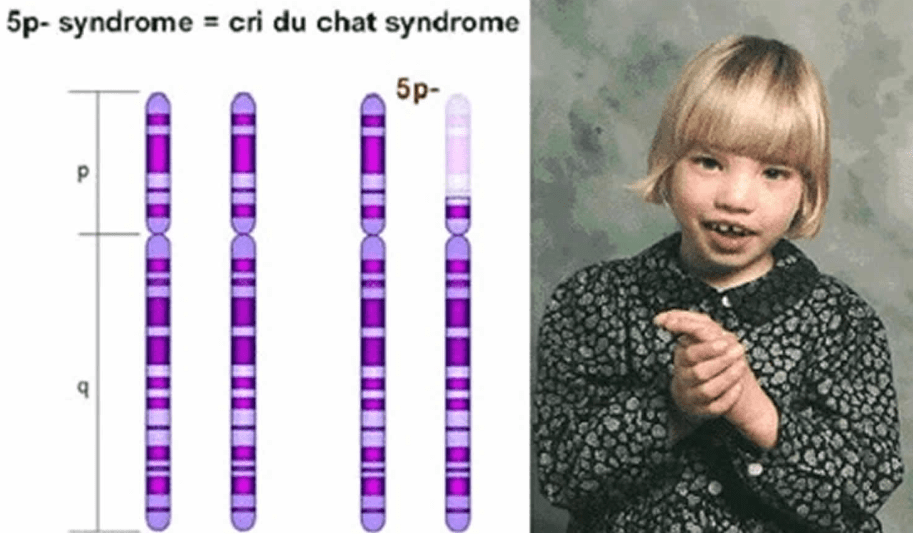
Segni e sintomi della sindrome di cri-du-chat
I sintomi della sindrome di cri-du-chat variano nel corso della vita e comprendono:
- Pianto acuto e monotono, che scompare entro i primi mesi di vita;
- Basso peso alla nascita;
- Microcefalia (testa piccola);
- Ipotonia (tono muscolare debole);
- Malformazioni cardiache, renali o intestinali;
- Malformazioni facciali che comprendono a loro volta:
- Occhi molto distanziati tra di loro (ipertelorismo);
- Volto arrotondato;
- Piega cutanea che copre l’angolo interno dell’occhio (epicanto);
- Bocca con angoli diretti verso il basso e l’esterno;
- Palato stretto;
- Mandibola di dimensioni ridotte (micrognazia).
- Ritardo nello sviluppo psicomotorio e cognitivo che comprende:
- Iperattività;
- Comportamento autolesionistico;
- Movimenti ripetitivi;
- Attaccamento ossessivo agli oggetti;
- La comprensione del discorso è migliore della loro capacità di esprimere o comunicare.
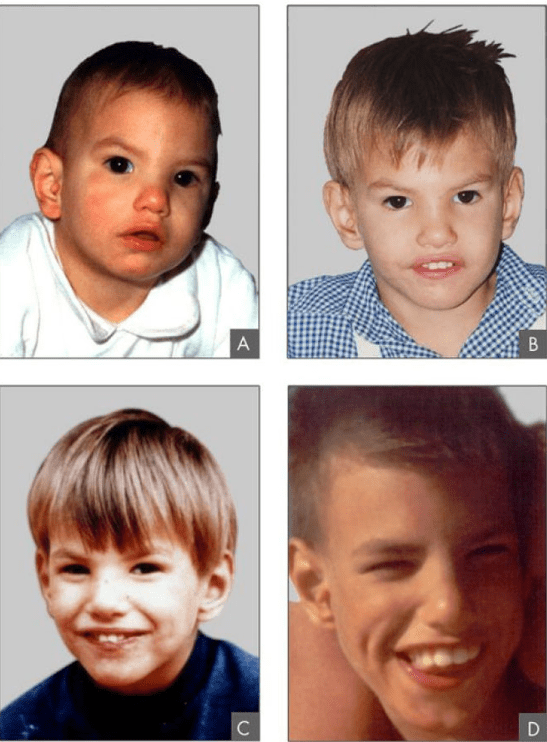
Epidemiologia
La sindrome di cri-du-chat è poco frequente e ha un range di incidenza da 1:15.000 a 1:50.000 nei neonati nati vivi. L’incidenza nelle femmine è leggermente superiore a quella dei maschi. Una volta che il bambino ha superato i primi anni di vita, la prognosi è buona e il livello di mortalità è basso.
Diagnosi per la sindrome cri-du-chat
La diagnosi per la sindrome di cri-du-chat può essere effettuata sia nel periodo prenatale che postnatale.
Nel periodo prenatale è diagnosticabile tramite:
- amniocentesi, dove sarà visibile la delezione del cromosoma 5;
- attraverso le ecografie sono osservabili le anomalie strutturali.
La diagnosi postnatale viene effettuata sulla base di risultati clinici. Se è presente un sospetto clinico, viene effettuata una conferma diagnostica tramite l’analisi del cariotipo. Ulteriori test specifici, sono:
- FISH identificherà le anomalie strutturali cromosomiche;
- Arrey-CGH (ibridazione genomica comparativa);
PCR quantitativa (reazione a catena della polimerasi)
Terapia per la sindrome di cri-du-chat
Ad oggi, non esiste una terapia specifica per i pazienti affetti dalla sindrome di cri-du-chat. Tuttavia, è possibile migliorare la prognosi attraverso interventi riabilitativi ed educativi individualizzati. I bambini possono essere seguiti da logopedisti, fisioterapisti e terapisti occupazionali. La fisioterapia, dovrebbe iniziare nelle prime settimane di vita, se i bambini hanno difficoltà ad aspirare e deglutire. I pazienti hanno spesso sordità neurosensoriale; pertanto, gli esami audiometrici dovrebbero essere eseguiti in tutti i bambini. Altri trattamenti utili includono la chirurgia, per correggere specialmente le anomalie cardiache, una dieta e routine salutare.
Fonti
- Genetica un approccio molecolare Russell terza edizione ISBN 9788871925882
- Ajitkumar A, Jamil RT, Mathai JK. Cri Du Chat Syndrome. 2022 Oct 7. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan–. PMID: 29494067.
- Liverani ME, Spano A, Danesino C, Malacarne M, Cavani S, Spunton M, Guala A. Children and adults affected by Cri du Chat syndrome: Care’s recommendations. Pediatr Rep. 2019 Feb 26;11(1):7839. doi: 10.4081/pr.2019.7839. PMID: 30838120; PMCID: PMC6397997.
- Cerruti Mainardi P. Cri du Chat syndrome. Orphanet J Rare Dis. 2006 Sep 5;1:33. doi: 10.1186/1750-1172-1-33. PMID: 16953888; PMCID: PMC1574300
- https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=IT&Expert=281
Fonti immagini
- Figura in evidenza: https://emedicine.medscape.com/article/942897-overview
- Figura 1: https://medlineplus.gov/genetics/condition/syndrome/#references
- Figura 2: https://healthjade.com/-syndrome/
- Figura 3: https://en.wikipedia.org/wiki/File