Descrizione
L’epilessia (dal greco ἐπιληψία, “essere preso, colto di sorpresa“) è un disturbo neurologico cronico che riguarda il sistema nervoso centrale in cui si verifica un’alterazione dell’attività neuronale (Fig.1), cioè delle cellule nervose deputate alla trasmissione elettro-chimica degli impulsi. Ciò causa convulsioni, crisi epilettiche, comportamenti insoliti e talvolta perdita di coscienza. In rari casi questi eventi possono vere una breve durata, tanto da passare quasi inosservati, ma possono prolungarsi per lunghi periodi.

Cause
Qualunque elemento di disturbo della normale attività neuronale quali patologie, traumi, o alterazioni dello sviluppo a livello cerebrale, può dare origine alle crisi epilettiche. Si stima che il 50% delle crisi che solitamente si verificano non riconoscono una causa nota: queste forme sono conosciute come epilessie primarie o idiopatiche. Il restate 50%, invece, sono conosciute come epilessie secondarie e sono riconducibile a vari fattori, tra cui:
- fattori genetici: alcuni tipi di epilessia sono ereditari. I ricercatori hanno stimato che siano circa 500 i geni che potrebbero essere legati allo sviluppo di questa condizione.
- traumi cerebrali: causati, ad esempio, da incidenti stradali o ad altre lesioni traumatiche di diverso tipo.
- patologie cerebrali: tumori cerebrali ed ictus, ad esempio, possono generare epilessia.
- lesioni prenatali: prima della nascita il feto è particolarmente sensibile a danni cerebrali indotti da svariati fattori, (un’infezione a carico della madre, una scarsa nutrizione o una carenza di ossigeno, ad esempio). Questi danni cerebrali possono successivamente indurre epilessia o paralisi cerebrale.
- malattie infettive: meningite, AIDS ed encefalite virale ne sono alcuni esempi.
Altre possibili cause possono essere legate a patologie degenerative (come il morbo di Alzheimer), mancanza di sonno o alterazioni metaboliche, iperosmolarità o iposmolarità.
Crisi epilettiche
L’epilessia è contraddistinta dalla cronica comparsa di crisi epilettiche indotte da un’eccessiva attività dei neuroni cerebrali, la cui scarica energetica è registrabile all’elettroencefalogramma. Si genera, dunque, una condizione convulsiva. Il paziente manifesta modificazioni comportamentali associate alla comparsa di svariati sintomi, ad esempio dalla comparsa di strane sensazioni/emozioni alle crisi convulsive e alla perdita di coscienza.
Classificazione delle crisi epilettiche
In base alla sede nella quale si manifesta l’attività cerebrale anomala distinguiamo crisi focali e crisi generalizzate.
Crisi focali
Le crisi focali, o parziali, sono caratterizzate da brusche scariche delle cellule nervose, originate in un solo emisfero cerebrale. Queste si dividono in:
- crisi focali semplici: non causano perdita di coscienza. Generano la comparsa di crisi visive e acustiche, crisi motorie, crisi somatosensoriali, crisi dismnesiche, affettive e cognitive.
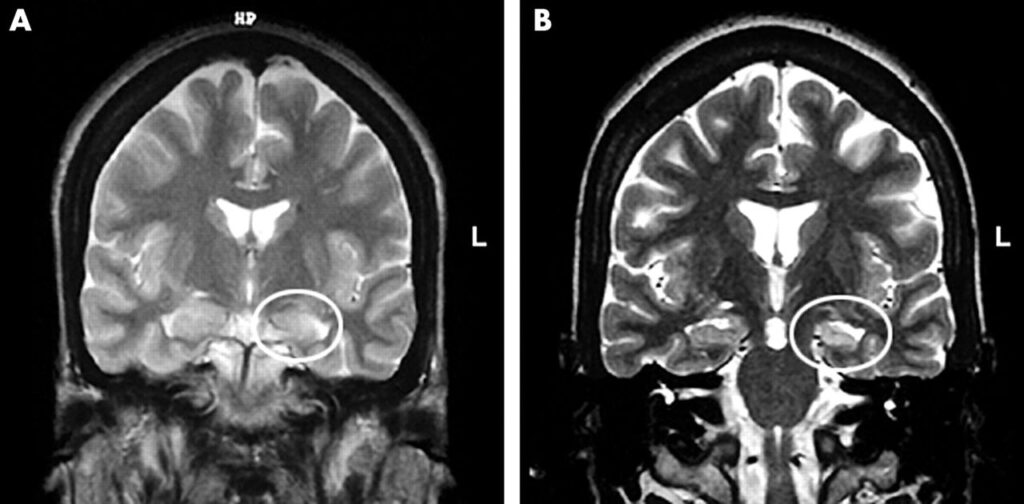
- crisi focali complesse (o discognitive): causano perdita di coscienza. Il paziente appare confuso ed esplica crisi psicomotorie. Sono tipiche dei focolai della corteccia dei lobi temporali o frontali (Fig.2).

Crisi generalizzate
Le crisi generalizzate sono dovute a un’alterazione dell’attività neuronale di entrambi gli emisferi cerebrali. Queste crisi rappresentano il 40% di tutte le sindromi epilettiche, e hanno generalmente un’eziologia genetica. Esistono sei tipi di crisi generalizzate:
- crisi di assenza: sono denominate anche “piccolo male” e sono caratterizzate da una rapida perdita di coscienza (da 4 a 20 sec) che colpisce frequentemente i bambini. Si manifesta con arresto improvviso dell’attività, sguardo fisso e spesso rotazione in alto degli occhi. Le assenze possono essere semplici (solo perdita di coscienza) o complesse (con contrazioni cloniche, ipotonia muscolare, ipertonia dei muscoli assiali).
- crisi cloniche: sono associate a movimenti muscolari bruschi e coinvolgono solitamente braccia, viso e collo.
- convulsioni toniche: caratterizzate da irrigidimento dei muscoli e coinvolgono i muscoli di braccia, schiena e gambe.
- crisi miocloniche: si manifestano solitamente come brevi torsioni o movimenti bruschi e improvvisi di braccia e gambe.
- convulsioni atoniche: determinano una perdita del tono muscolare, che può indurre un improvviso collasso o caduta del soggetto.
- crisi tonico-cloniche: sono denominate anche “grande male” e rappresentano il tipo più grave di crisi epilettica. Hanno una durata di circa 5-10 minuti e sono caratterizzate da una fase di contrazione intensa che riguarda tutto il corpo, una fase caratterizzata da convulsioni e una fase di risoluzione caratterizzata da respirazione rumorosa e spesso perdita di urine. Il paziente non conserva alcun ricordo della crisi.
Diagnosi
I sintomi dell’epilessia focale possono essere confusi con quelli di altri disturbi neurologici, quali emicrania, la narcolessia e altri disturbi mentali. Dunque il punto di partenza per diagnosticare l’epilessia consiste in un attento esame dei sintomi, un’accurata descrizione di ciò che avviene nei momenti immediatamente precedenti la comparsa della crisi, durante il suo sviluppo e al suo termine. Inoltre è importante sottoporre il paziente a diversi esami di approfondimento.
La Risonanza Magnetica Nucleare (MRI), esame cardine per la diagnosi ed il trattamento di pazienti con epilessia, sfrutta onde radio e potenti magneti per ricreare un’immagine dettagliata del cervello, consentendo così l’identificazione di eventuali lesioni o anomalie cerebrali (Fig.3). L’MRI funzionale (fMRI) misura le variazioni del flusso sanguigno che si verificano in particolari regioni funzionali del cervello, come quelle di linguaggio e movimento.
Altri test diagnostici
L’elettroencefalogramma (EEG), test più comunemente utilizzato, valuta l’attività elettrica del cervello (Fig.4) registrandone le onde, che spesso risultano alterate nei soggetti con epilessia. Nonostante l’accuratezza di questo test, alcuni soggetti continuano a mostrare onde cerebrali normali anche dopo la comparsa delle crisi.

La Tomografia Computerizzata (TC) sfrutta i raggi X al fine di ottenere sezioni trasversali del cervello. Attraverso questa tecnica si può identificare la presenza di anomalie cerebrali che possono innescare le crisi epilettiche, quali tumori, emorragie e cisti.
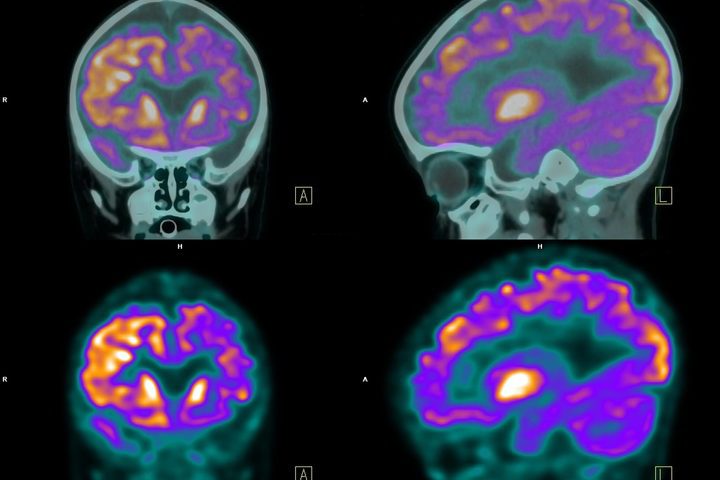
La Tomografia a emissione di positroni (PET) sfrutta un materiale radioattivo a basso dosaggio che viene iniettato per via endovenosa per visualizzare le aree cerebrali attive e identificare le anomalie (Fig.5).

Epilessia e mutazioni genetiche
La nota rivista Nature Genetics ha reso pubblico uno studio condotto da un team di ricercatori finanziati dall’UE, il quale ha scoperto che una mutazione sul cromosoma 15 (Fig.6) è legato ad una forma comune di epilessia. L’epilessia è una malattia neurologica molto diffusa e circa la metà dei casi totali presentano una forte componente genetica: finora sono stati scoperti 20 geni collegati a questa malattia collegati a forme epilettiche molto rare.
Gli scienziati hanno analizzato il DNA di oltre mille persone affette da epilessia generalizzata idiopatica (IGE) e lo hanno confrontato con il DNA di oltre 3.500 soggetti sani. Hanno rilevato che una piccola sezione del cromosoma 15 mancava nell’1% dei malati di IGE, mentre nessuna delle persone sane esaminate possedeva questa delezione. Inoltre ricerche precedenti hanno collegato la stessa delezione sul cromosoma 15 a disabilità intellettuali, schizofrenia e/o altre sindromi neuropsichiatriche. La sezione mancante di DNA contiene almeno sette geni, tra cui uno che codifica la proteina CHRNA7 (recettore neuronale nicotinergico dell’acetilcolina), la quale regola i segnali tra le cellule nervose.

Trattamenti dell’epilessia
La comparsa delle convulsioni può essere evitata attraverso l’utilizzo di farmaci anti-epilettici ma, in alcuni casi, questi potranno solo ridurre frequenza o intensità delle crisi.
Recenti studi hanno affermato che le crisi ripetute (o prolungate) possono portare allo sviluppo di una epilessia “cronica” (Fig.7). Ciò si verifica perché le connessioni fra i neuroni possono riorganizzarsi in modo scorretto: il fenomeno è riconosciuto come plasticità reattiva.
Sulla nota rivista Annals of Neurology è stato pubblicato uno studio sperimentale condotto nel Centro di Neuroscienze dell’Università di Helsinki, il quale dimostra il ruolo del mutamento nella funzione dell’acido gamma-amino-butirrico (GABA), il principale neurotrasmettitore inibitorio cerebrale, che genera una condizione di patologica riorganizzazione di circuiti neuronali, in grado di produrre una epilessia cronica. Normalmente i neuroni che hanno come neurotrasmettitore il GABA svolgono un’azione inibitoria, di controllo, in grado di evitare una eccessiva eccitabilità degli altri neuroni. Dopo crisi epilettiche prolungate o ripetute i neuroni GABA possono modificare la loro attività e di conseguenza aumentare, anziché inibire, l’eccitabilità di altri neuroni e quindi facilitare nuove e dannose connessioni neurali.
Meccanismo d’azione dei farmaci anti-epilettici
Nell’epilessia si instaura uno squilibrio tra le principali vie di neurotrasmissione: la via glutammatergica (eccitatoria) e quella GABAergica (inibitoria). In questo squilibrio, che porta a crisi epilettica, giocano un ruolo determinante i meccanismi di regolazione della soglia, della durata e della propagazione dell’evento convulsivo: i canali ionici voltaggio-dipendenti di Na+,K+,Ca2+ e i canali ionici ligando-dipendenti di glutammato e GABA.
L’azione dei farmaci antiepilettici (Fig.8) consiste in:
- Inattivazione dei canali del Na+ voltaggio-dipendenti
- Inibizione dei canali del Ca2+ voltaggio-dipendenti di tipo T
- Potenziamento della trasmissione GABAergica

Principali farmaci anti-epilettici
I farmaci che potenziano l’azione del GABA più comuni sono: il fenobarbital, valproato, progabide e gabapentin. Tra i farmaci che, invece, agiscono nella soppressione dei canali per il sodio citiamo: fenitoina, lamotrigina e carbamazepina. Tra i farmaci inibitori dei canali del calcio c’è il levetiracetam. Ovviamente la scelta del farmaco più appropriato può risultare complessa, poiché si basa su diversi fattori quali il tipo di epilessia riscontrata, la frequenza delle convulsioni, l’età.
Anna Maria Musto
Fonti
- Nazim Kourdougli PhDChristophe Pellegrino PhDJuho‐Matti Renko PhDStanislav Khirug PhDGeneviève Chazal PhDTiina‐Kaisa Kukko‐Lukjanov PhDSari E. Lauri PhD, “Depolarizing γ‐aminobutyric acid contributes to glutamatergic network rewiring in epilepsy”. Annals of Neurology, 2017.
- Carlos A. M. Guerreiro, “Epilepsy: Is there hope?”. Indian Journal of Medical Research, 2016.
- Ingo Helbig, Heather C Mefford,Thomas Sander “15q13.3 microdeletions increase risk of idiopathic generalized epilepsy“. Nature Genetics, 2009.
- Eric B Geller, Tara L Skarpaas, Robert E Gross, Robert R Goodman, Gregory L Barkley, Carl W Bazil, Michael J Berg, Gregory K Bergey, Sydney S Cash, Andrew J Cole, Robert B Duckrow, Jonathan C Edwards, Stephan Eisenschenk, James Fessler, Nathan B Fountain, Alicia M Goldman, Ryder P Gwinn, Christianne Heck, Aamar Herekar, Lawrence J Hirsch, Barbara C Jobst, David King-Stephens, Douglas R Labar, James W Leiphart “Brain-responsive neurostimulation in patients with medically intractable mesial temporal lobe epilepsy“, 2017.
Un articolo completo, ben organizzato e integrato con un valido repertorio iconografico !