Descrizione
Si ritiene che i disturbi legati alla Corea di Huntington si conoscano sin dal medioevo, ma le sue particolarità e le sue cause sono rimaste sconosciute per molto tempo.
Venne dapprima identificata con il nome di “corea”, per i caratteristici movimenti a scatti e simil- danzanti, per poi essere chiamata “corea ereditaria” e “corea cronica progressiva”.
Fu il medico statunitense George Huntington, nel 1872, a delineare i primi aspetti della patologia, nel suo articolo intitolato “On Chorea”.
La definì come un disordine di tipo ereditario, con una tendenza alla infermità mentale e con una insorgenza tardiva.
Epidemiologia
La Corea di Huntington è presente a livello globale in 5-10 casi ogni 100.000 persone, ma la distribuzione geografica mostra variazioni in base alle etnie e alle migrazioni.
Non ci sono diversità sostanziali di incidenza tra uomini e donne. Sono visibili, invece, tassi di incidenza differenti per quanto riguarda popoli europei e resto del mondo.
In Europa, soprattutto nella parte occidentale, l’incidenza è in media di 3-7 casi su 100.000 persone. Popolazioni asiatiche o africane, invece, mostrano un’incidenza di 1 caso su 1.000.000.
In popolazioni venezuelane, invece, si arriva ad una incidenza di 7000 casi su 1.000.000 di persone. Scozia, Galles, Svezia e Tasmania mostrano anch’esse un’incidenza elevata, suggerendo come possibile causa l’effetto del fondatore in una zona di emarginazione geografica.
La Corea di Huntington è una malattia autosomica dominante, quindi la probabilità di ereditare il gene da un genitore malato è del 50%. Difficilmente il gene è presente in entrambi i genitori, ma in quei rari casi la probabilità di patologia arriva anche al 100% nella generazione successiva. Inoltre, se il gene è ereditato dalla parte paterna, l’anticipazione genetica risulta essere più grave: poiché durante la spermatogenesi si verifica l’ampliamento della mutazione.
La Corea di Huntington è ad insorgenza tardiva, in quanto si riscontra intorno ai 40-50 anni di età, con una lunga degenza che arriva anche ai 25 anni. Nelle generazioni successive l’insorgenza può scendere sotto i 20 anni di età, ed avere una degenza ben più corta.
Patogenesi e fattori di rischio
Il morbo di Huntington è una malattia neurodegenerativa del Sistema Nervoso Centrale, autosomica dominante e ad insorgenza tardiva. La presenza di disturbi a livello motorio, psichiatrico e cognitivo descrivono un quadro patologico davvero molto rilevante.
La comparsa della patologia risulta essere strettamente collegata a sequenze nucleotidiche, presenti nel gene specifico, ripetute in quantità elevata e ciò si ripercuote sulla quantità di proteina anomala prodotta.
L’aumento del numero delle sequenze nelle generazioni successive è un fenomeno che prende il nome di “anticipazione genetica”, e ciò comporta aumento della gravità dei sintomi ed una comparsa sempre più prematura dell’insorgenza.
La proteina anomala è di fatto ubiquitaria nell’organismo, ma la degenerazione è presente maggiormente nel cervello.
Si ritiene che interagisca con oltre cento altre proteine, svolgendo svariate funzioni, ma, nonostante ciò, il comportamento della proteina mutata non è ancora completamente chiaro.
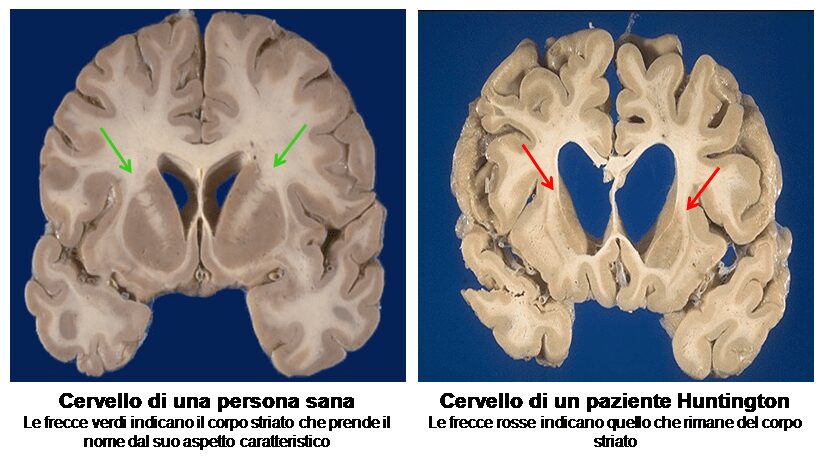
La certezza, però, è che risulta essere estremamente dannosa per alcuni tipi di cellule, tra cui quelle del corpo striato, detto anche nucleo della base o nucleo caudale. Quest’ultime risultano subire il danno precoce più vistoso, in quanto la proteina anomala determinerà la morte cellulare.
Con l’avanzare dello stato patologico, il danno intaccherà i neuroni presenti nella corteccia, più in superficie, portandoli a morte. Essendo progressiva, pian piano tutto il cervello andrà incontro ad atrofia, evidenziando, quindi, deficit cognitivi e motori sempre maggiori.
Descrizione anomalia genetica
Si tratta di una patologia monogenica, in quanto è solamente un gene a presentare la mutazione responsabile.

Nello specifico, il gene IT15 situato nel cromosoma 4 (locus 4p16.3), presenta una particolare sezione genica ripetuta in misura maggiore del normale. La Corea di Huntington è quindi una delle malattie da espansione di triplette, fenomeno genetico molto particolare:
una sequenza di tre basi azotate, chiamata tripletta, è ripetuta molte più volte, determinando una marcata situazione patologica.
Nel morbo di Huntington la sequenza Citosina–Adenina–Guanina (CAG) è presente più di 36 volte. Maggiore sarà il numero di ripetizioni e maggiori saranno gli impatti patologici, aggravando la sintomatologia e l’esordio della malattia.
La presenza di un numero maggiore di triplette determina la produzione di un prodotto genico errato, ovvero il gene produrrà una proteina, chiamata Huntingtina (HTT), anomala.

Vanno elencati i range patologici:
- Meno di 26 ripetizioni il soggetto risulterà sano;
- Tra 27 e 35 ripetizioni il soggetto sarà sano ma avrà un percentuale elevata di rischio per la progenie;
- Tra 36 e 39 ripetizioni il soggetto presenterà un range premutato, ovvero il soggetto è fenotipicamente sano con possibilità di sviluppare la malattia, e predisposto alla produzione di gameti con la mutazione piena;
- Più di 40 ripetizioni il soggetto avrà un range di mutazione piena. Manifesta insorgenza tardiva della patologia, ed inoltre, tanto è più grande il numero di espansioni della tripletta, tanto più precoce sarà l’età di insorgenza.
Caratteristiche cliniche e complicanze
I primi sintomi a manifestarsi sono solitamente quelli fisici rispetto ai cognitivi e psichiatrici.
In ogni caso ogni sintomo ha una entità, un esordio ed uno sviluppo del tutto soggettivo tra le persone.
Tra i fisici più caratteristici abbiamo dei movimenti a scatti ingestibili e casuali, spesso preceduti da lievi movimenti involontari. Con la progressione patologica compariranno poi rigidità o posture anomale. Via via, ogni funzione motoria verrà meno.
Anche le capacità cognitive andranno peggiorando con il processo patologico. Deficit mnemonici, demenza progressiva, manifestazioni neuropsichiatriche ed azioni compulsive saranno sempre più presenti.
In una fase avanzata, l’HTT mutante presente nei tessuti periferici comporterà la presenza di atrofia muscolare, insufficienza cardiaca, atrofia testicolare ed osteoporosi.
Diagnosi
Nel 99% dei casi la diagnosi di Corea di Huntington è confermata da test genetici in seguito alla comparsa di alcuni sintomi. I test confermeranno la presenza della ripetizione delle triplette. Inoltre, il morbo di Huntington può presentarsi anche in seguito a patologie non genetiche come ictus o infezioni da HIV. Esami di immagini biometrico, tra le quali la tomografia computerizzata o la risonanza magnetica forniscono la possibilità di visionare l’atrofia del nucleo caudato. Ma questo aspetto non rientra nella diagnosi precoce della malattia, quindi sono da ritenersi esami strumentali successivi. Il test genetico permette la diagnosi precoce rispetto alla comparsa dei sintomi fisici, ed anche la diagnosi prenatale.
Trattamento e prevenzione
Al momento non esiste alcuna cura per la Corea di Huntington, ma sono presenti dei farmaci capaci di diminuire la gravità di alcuni sintomi. Si può ricorrere a terapie di aiuto nella gestione della malattia, tra cui fisioterapia specifica formulata dalla European Huntington’s Disease Network. Oppure l’utilizzo della terapia occupazionale o della logopedia.
Tra i farmaci utilizzati per alcuni effetti benefici abbiamo la tetrabenazina, le benzodiazepine e altri della famiglia dei neurolettici. Inoltre, per i sintomi psichiatrici, possono essere utilizzati farmaci in uso per depressione e per alcuni fenomeni di psicosi.
Fonti
- https://it.wikipedia.org/wiki/George_Huntington
- https://it.wikipedia.org/wiki/Malattia_di_Huntington
- https://www.sciencedirect.com/science/article/abs/pii/S163470721889403X
- https://www.cell.com/molecular-cell/fulltext/S1097-2765(04)00545- 3?_returnURL=https%3A%2F%2Flinkinghub.elsevier.com%2Fretrieve%2Fpii%2FS10972765040054 53%3Fshowall%3Dtrue
- https://www.huntington-onlus.it/la-malattia-di-huntington/cose-la-malattia-di-huntington/
- https://pubmed.ncbi.nlm.nih.gov/17240289/
- https://www.sciencedirect.com/science/article/abs/pii/B9780444520142000252?via%3Dihub
- https://www.medimagazine.it/nuovo-possibile-trattamento-per-la-malattia-di-huntington/
- https://www.medimagazine.it/nuovo-trattamento-ha-ridotto-lattivita-del-gene-responsabile-della-malattia-di-huntington/
- https://it.wikipedia.org/wiki/Huntingtina
- https://www.repubblica.it/2009/01/sezioni/scienze/cervello/causa-corea-di-huntington/causa-corea-di-huntington.html