Cos’è la proteomica ?
Prima di addentrarci nello studio vero e proprio, iniziamo definendo il concetto di proteomica, disciplina nata fra gli anni 90 e i primi anni 2000 facente parte delle scienze omiche, utilizza tecnologie di analisi che consentono la produzione di informazioni (dati), in numero elevato, utili per la descrizione e l’interpretazione del sistema biologico studiato; infatti nasce nell’era post genomica con l’avvento delle tecniche di sequenziamento. Non a caso il termine “omico” deriva dal greco e significa “tutto”.
Quindi la proteomica la possiamo definire come lo studio su larga scala dell’insieme delle proteine originate da un genoma. A differenza del genoma che è uguale in ogni singola cellula di un organismo, il proteoma si presenta in modo diverso a seconda del tipo di cellula e dello stato del ciclo cellulare; ad esempio prendendo una cellula dell’epidermide umano e una nervosa, ambedue avranno gli stessi 21.000 geni (circa), ma alcuni si esprimono nella cellula nervosa e non in quella dell’epidermide e viceversa. Questo caratterizza il fenotipo di queste due tipi di cellula in virtù della regolazione genica che modula l’espressione dei geni e di conseguenza delle proteine, causando un differenziamento dell’espressione del proteoma.
Da questo piccolo esempio abbiamo capito una caratteristica importante del proteoma ossia la sua dinamicità. Il proteoma quindi quindi la capacità di cambiare e diversificarsi in virtù della regolazione genica, ma non solo anche per: stimoli esterni e ormonali, mutazioni, fenomeni di splicing (eliminazione parte del genoma non codificante), momenti diversi del ciclo cellulare e modificazioni post traduzionali (glicosilazione, fosforilazione ecc); proprio su quest’ultime ci focalizzeremo nel seguito di questo articolo (Analisi del fosfoproteoma delle cellula HeLa).
A questo punto se vogliamo riassumere in poche parole qual è l’obbiettivo principale di questa scienza prossimo dire che:
La sfida a lungo termine della proteomica è enorme: stabilire le identità, le quantità, le strutture e le funzioni dei complementi completi di proteine, e caratterizzare come queste proprietà variano nei diversi contesti cellulari
da un articolo di Tyers & Mann del 2003
Il dynamic range
L’obiettivo di studiare le proteine su larga scala incontra molte inside a livello analitico, fra le più importanti c’è quella del dynamic range che è legato al fatto che l’abbondanza relativa delle proteine all’interno di un organismo è molto diversa: possiamo avere proteine molto abbondanti (milioni di copie) e proteine molto poco abbondanti (unità di copie). Per comprendere il concetto di dynamic range basti pensare alla composizione proteica di un campione di plasma (componente non corpuscolare del sangue) umano, nonostante sia un sistema estremamente più semplice di quanto possa essere una cellula: anche in questo caso, tuttavia, le abbondanze relative proteiche sono evidentemente diverse: albumina nell’ordine dei mg/mL, gastrina nell’ordine dei ng/mL, citochine (importanti nella risposta immunitaria) nell’ordine dei pg/mL, dato che alcune proteine svolgono la loro funzione a basse contrazioni, mentre altre necessitano di un espressione più elevata.
Per arginare tale limiti si usano una combinazione di strumenti che cercano di aumentare la sensibilità e risoluzione dell’analisi proteomica, come ad esempio l’utilizzo di elettroforesi 2D-SDS-PAGE e dello spettrometro di massa.
La proteomica, trovano applicazione in varia ambiti della ricerca come ad esempio nello studio e ricerca di biomarker per la diagnosi precoce di tumori, dato che questo tipo di malattie fanno variare il proteoma della linea cellulare interessata.
Studio proteomico di modificazioni post-traduzionali
Le modificazioni post-traduzionali costituiscono un fattore centrale nel determinare la diversità e la complessità del proteoma. È molto importante studiarle, in quanto presuppongono una variazione nell’effetto della proteina stessa. Lo studio delle modificazioni post-traduzionali richiede, in aggiunta alla spettrometria di massa, una combinazione di tecniche (possibilmente di arricchimento) più o meno elaborate per l’identificazione e la separazione delle proteine recanti una specifica modificazione, tra queste:
- la cromatografia di affinità
- l’impiego di anticorpi specifici contro la modificazione
- l’uso di radioisotopi
- l’aggiunta di tags covalenti (marcatori) per il riconoscimento di specifici gruppi chimici.
Le analisi proteomiche che interessano le modificazioni post traduzionali richiedono più materiale, rispetto ai classici studi di proteomica, dato che si studia sottogruppo di proteine caratterizzate non solo a livello del tipo di modificazione, ma anche alla localizzazione della modificazione (ad esempio, se la proteina è fosforilata, occorre conoscere dove si posizionano i gruppi fosfato per sapere come influenza la funzione delle stessa in virtù del sito di legame). Esistono moltissimi tipi di modificazioni post-traduzionali, in questo articolo come già anticipato ne vedremo un caso specifico; cioè il fosfoproteoma di cellule HeLa.
Fosfoproteoma di cellule HeLa: lo studio
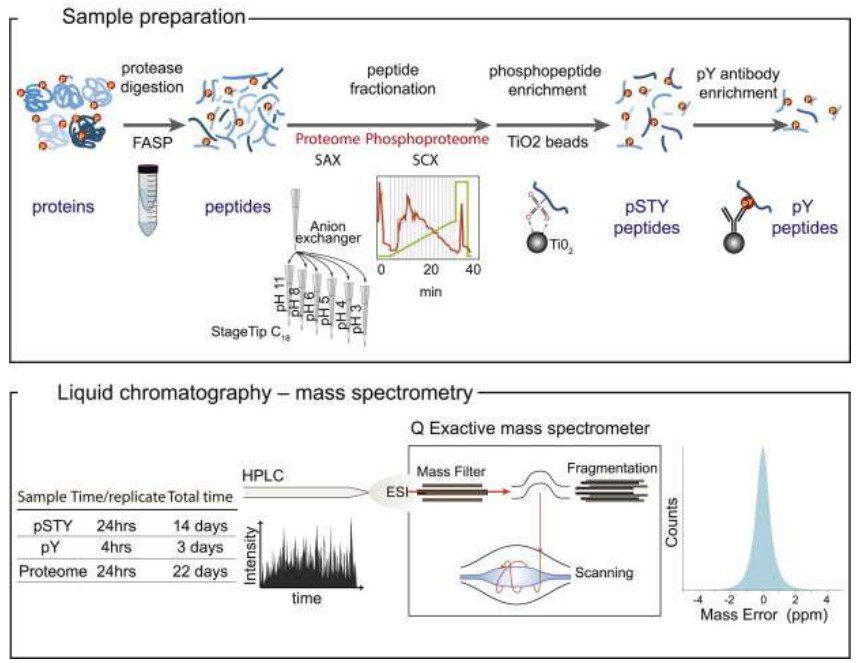
Questo studio è stato condotto su cellule HeLa, che devono il loro nome a Henrietta Lacks che, nel 1951 è morta di un tumore all’utero: sono cellule che costituiscono un modello molto utilizzato molto utilizzato negli ultimi 60-65 anni, dato che hanno una derivazione umana; essendo tumorali sono in grado di replicarsi senza nessuna limitazione temporale in laboratorio (molti aspetti regolativi sono andati persi). È proprio su questa linea cellulare che, nel 2014, un gruppo di ricercatori ha eseguito analisi relativamente alla composizione del fosfoproteoma, con un livello di sensibilità elevato (fosfoproteoma ultraprofondo) rispetto ad altri studi di proteomica, allo scopo di approfondire la conoscenza rispetto alla quantità, alla percentuale di una proteina, alla posizione di fosforilazione e così via, cercando di superare il problema del dynamic range.
Protocollo sperimentale del fosfoproteoma di cellule HeLa:
- Realizzazione di un estratto cellulare di cellule HeLa coltivate in vitro
- trattamento dell’estratto con una soluzione contenente le proteasi tripsina e lysC, per fornire un grande numero di peptidi
- Prearricchimento dei peptidi con tecniche di cromatografia bidimensionale SAX (cromatografia a scambio anionico), per avere una mappatura del proteoma
- Aarrichimento tramite SCX (cromatografia a scambio cationico), per selezionare i fosfopeptidi
- Ulteriore arricchimento utilizzando cromatografia MOAC (con resine derivatizzate al biossido di titanio) per un ulteriore purificazione dei fosfopeptidi
- Trattamento di arricchimento dei fosfopeptidi con anticorpi contro la fosfotirosina, per cui verrà selezionata solo una porzionale, dell’intero fosfoproteoma
- Analisi dei peptidi utilizzando cromatografia liquida HPLC accoppiata a uno spettrometro di massa. Questo consente di caratterizzare i peptidi, di assegnarli alla proteina da cui derivano, di identificare qual è il residuo fosforilato (a livello di struttura primaria della proteina), e di comprendere la percentuale di fosforilazione della proteina
Risultati
Dall’analisi del fosfoproteoma delle cellula HeLa sono state visualizzate un totale di:
- 10800 proteine (circa la metà delle proteine possibili sulla base del numero di geni umani, 21000 (circa), per possibili eventi di riarrangiamento genetico dei geni delle cellule tumorali, per assenza di espressione o, ancora, per un’espressione molto bassa, che ci impediva di distinguere i peptidi dal rumore di fondo)
- delle 10800 ben 7800 sono fosforilate (circa il 70% del totale)
Dallo studio si identificano 51000 fosfopeptidi diversi e, per molti di questi, circa 38000, anche fosfosito (sito di fosforilazione).
Si è visto che la fosforilazione interessava:
- 80% i residui di serina
- 20% i residui di treonina
- 0,4% i residui di tirosina
Conclusioni sul fosfoproteoma di cellule HeLa
Da questo studio si può evincere l’importanza di questo tipo di modificazione post-traduzionale; infatti se si osserva il genoma umano ben 500 geni codificano per protein-chinasi, cioè gli enzimi coinvolti nel processo di fosforilazione, e da altri studi proteomici si è evidenziato che ben il 40% delle proteine totali presenta fosforilazioni, modificazione che coinvolge soprattutto serine, tirosine e treonine (substrati preferiti dall’enzima chinasi).
Questa modificazione modificazione ha un importanza molto elevata nei processi regolativi cellulari; ne è un esempio la proteina p-53, coinvolta nella regolazione del ciclo cellulare, che presenta ben 18 siti di fosforilazione, ed in base a dove e quanto avviene la modificazione, essa assumerà funzioni diverse.
Quindi prossimo afferma che l’analisi del fosfoproteoma delle cellula HeLa, ha permesso di ampliare le conoscenze di una delle modificazioni post-traduzionali più presente in eucarioti
Fonti
- Tyers, M. & Mann, M. (2003) “From genomics to proteomics” , Nature 422, pp. 193-197.
- Kolker E, Higdon R, Hogan JM. (2006) “Protein identification and expression analysis using mass spectrometry.” Trends Microbiol. 14, pp. 229-35.
- Sharma K, et al (2014) “Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling.” Cell Rep. 8, pp 1583-94.
Immagini
- figura 1: https://www.google.com/url?sa=i&url=https%3A%2F%2Fmicrobe.com%2Fproteomics-er%2F&psig=AOvVaw0jj5XxNZuwR6cobfRX0D49&ust=1676226553181000&source=images&cd=vfe&ved=0CBEQjhxqFwoTCKi3n9SMjv0CFQAAAAAdAAAAABAE
- figura 2: Sharma K, et al (2014) “Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling.” Cell Rep. 8, pp 1583-94.