La struttura delle proteine determina la funzione biologica
Quando guardiamo una cellula al microscopio o ne analizziamo l’attività elettrica o biochimica stiamo essenzialmente osservando proteine. Le proteine costituiscono la maggior parte della massa secca di una cellula. Esse non sono soltanto le unità di cui sono costituite le cellule, ma svolgono anche quasi tutte le funzioni cellulari. Così gli enzimi forniscono a una cellula le intricate superfici molecolari che promuovono le sue numerose reazioni chimiche. Proteine immerse nella membrana plasmatica formano canali e pompe che controllano il passaggio di piccole molecole dentro e fuori la cellula.
Altre proteine conducono messaggi da una cellula all’altra o agiscono come integratori di segnali che ritrasmettono serie di segnali dalla membrana plasmatica all’interno della cellula fino al nucleo. Altre ancora servono da minuscole macchine molecolari con parti in movimento: la chinesina, per esempio, muove organelli attraverso il citoplasma; la topoisomerasi può districare molecole annodate di DNA. Alcune, più specializzati, agiscono da anticorpi, tossine, ormoni, molecole anticongelanti, fibre elastiche, funi o fonti di luminescenza. Per poter sperare di comprendere come funzionano i geni, come i muscoli si contraggono, come i nervi conducono elettricità, come si sviluppa un embrione o come funzionano i nostri corpi, dobbiamo raggiungere una conoscenza profonda delle proteine.
- Leggi anche “Batteri, lieviti ed energie rinnovabili trasformano l’anidride carbonica in proteine per uso alimentare“
La forma e la struttura delle proteine
Da un punto di vista chimico le proteine sono di gran lunga le molecole strutturalmente più complesse e funzionalmente più sofisticate. Ciò probabilmente non è sorprendente, una volta che ci si rende conto che la struttura e la chimica di ciascuna proteina si sono sviluppate e raffinate in miliardi di anni di storia evolutiva. I calcoli teorici dei genetisti di popolazione rivelano che, in un arco di tempo evolutivo, è sufficiente un vantaggio selettivo sorprendentemente piccolo per causare la diffusione di una proteina, la cui sequenza è stata alterata in maniera casuale, in una popolazione di organismi.
Eppure, anche agli esperti, la notevole versatilità delle proteine può sembrare veramente stupefacente. In questo articolo considereremo il modo in cui la posizione di ciascun amminoacido nella lunga sequenza che dà origine a una proteina ne determini la forma tridimensionale. Useremo quindi questa conoscenza della struttura delle proteine a livello atomico per spiegare come la forma precisa di ciascuna proteina ne determini la funzione in una cellula.
La forma di una proteina è specificata dalla sua sequenza di amminoacidi
Nelle proteine esistono 20 tipi di amminoacidi, codificati direttamente dal DNA dell’organismo, ciascuno con proprietà chimiche diverse. Una proteina è costituita da una lunga catena di questi amminoacidi, ciascuno legato al suo vicino da un legame peptidico covalente. Le proteine sono perciò note anche come polipeptidi. Ciascun tipo di proteina ha una sequenza caratteristica di amminoacidi e ci sono parecchie migliaia di proteine diverse in una cellula. La sequenza ripetuta di atomi lungo il nucleo della catena polipeptidica viene chiamata ossatura polipeptidica. Attaccate a questa catena ripetitiva vi sono quelle porzioni degli amminoacidi che non sono coinvolte nella formazione di un legame peptidico e conferiscono a ciascun amminoacido le sue proprietà peculiari: le 20 diverse catene laterali degli amminoacidi. Alcune di queste catene laterali sono non polari e idrofobiche (“che temono l’acqua”), altre sono cariche negativamente o positivamente, alcune formano velocemente legami covalenti, e così via.

Gli atomi si comportano più o meno come se fossero sfere solide con un raggio definito (il raggio di van der Waals). L’impossibilità di sovrapposizione fra due atomi limita di molto i possibili angoli di legame in una catena polipeptidica. Questa restrizione e altre interazioni steriche riducono drasticamente la varietà di disposizioni tridimensionali (o conformazioni) degli atomi possibili.
Nonostante ciò, una lunga catena flessibile, come è una proteina, può ancora ripiegarsi in un numero enorme di modi. Il ripiegamento di una catena proteica è tuttavia ulteriormente limitato da molte serie differenti di deboli legami non covalenti che si formano fra parti diverse della catena. Questi coinvolgono atomi dell’ossatura polipeptidica, oltre ad atomi delle catene laterali degli amminoacidi. I legami deboli sono di tre tipi: legami idrogeno, attrazioni elettrostatiche e attrazioni di van der Waals. I singoli legami non covalenti sono 30-300 volte più deboli dei tipici legami covalenti che creano le molecole biologiche. Ma molti legami deboli possono agire in parallelo per tenere due regioni di una catena polipeptidica strettamente legate. In questo modo la stabilità di ciascuna forma ripiegata è determinata dalla forza combinata di un gran numero di questi legami non covalenti.



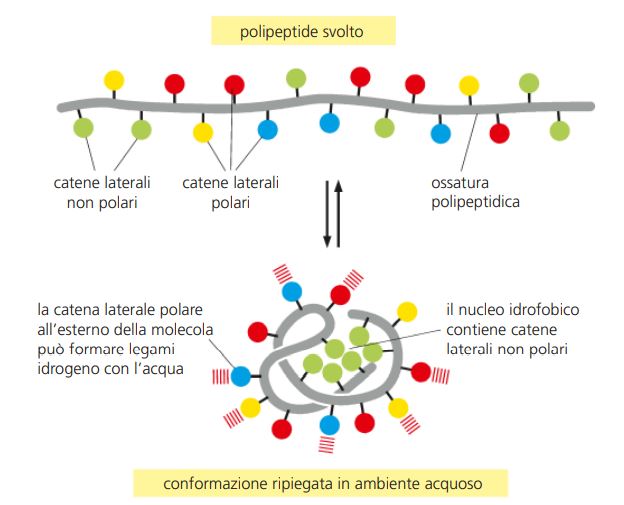
Una quarta forza debole ha un ruolo centrale nel determinare la forma di una proteina. Le molecole idrofobiche, comprese le catene laterali non polari di particolari amminoacidi, tendono a unirsi in un ambiente acquoso per ridurre al minimo i loro effetti che alterano la rete di legami idrogeno delle molecole d’acqua. Perciò, un fattore importante che governa il ripiegamento di qualunque proteina è la distribuzione dei suoi amminoacidi polari e non polari.
Le catene laterali non polari (idrofobiche) di una proteina – che appartengono ad amminoacidi quali fenilalanina, leucina, valina e triptofano – tendono a raggrupparsi nell’interno della molecola (proprio come goccioline d’olio idrofobiche si uniscono nell’acqua formando una goccia più grande). Ciò permette loro di evitare il contatto con l’acqua che le circonda all’interno di una cellula. Le catene laterali polari – come quelle che appartengono ad arginina, glutammina e istidina – tendono invece a disporsi vicino all’esterno della molecola, dove possono formare legami idrogeno con l’acqua e con altre molecole polari. Gli amminoacidi polari immersi all’interno della proteina in genere sono legati da legami idrogeno ad altri amminoacidi polari o all’ossatura polipeptidica.
Le proteine si ripiegano nella conformazione con l’energia più bassa
Come risultato di tutte queste interazioni la maggior parte delle proteine ha una struttura tridimensionale particolare, che è determinata dall’ordine degli amminoacidi nella sua catena. La struttura finale ripiegata, o conformazione, adottata da una catena polipeptidica è in genere quella che riduce al minimo l’energia libera. Il ripiegamento delle proteine è stato studiato in provetta usando proteine altamente purificate.
Una proteina può essere svolta, o denaturata, mediante trattamento con certi solventi, che distruggono le interazioni non covalenti che tengono insieme la catena ripiegata. Questo trattamento converte la proteina in una catena polipeptidica flessibile che ha perso la sua forma naturale. Quando il solvente denaturante viene rimosso la proteina spesso si ripiega spontaneamente, o rinatura, nella sua conformazione originale, il che sta a indicare che tutte le informazioni necessarie per specificare la forma tridimensionale di una proteina sono contenute nella sua sequenza di amminoacidi e questo è un punto cruciale per comprendere la biologia cellulare. La maggior parte delle proteine si ripiega in un’unica conformazione stabile. Tuttavia la conformazione spesso cambia leggermente quando la proteina interagisce con altre molecole nella cellula. Questo cambiamento di forma è spesso cruciale per la funzione della proteina.
Sebbene una catena proteica si possa ripiegare nella sua conformazione corretta senza aiuto esterno, il ripiegamento delle proteine in una cellula vivente è spesso assistito da proteine speciali chiamate chaperoni molecolari. Queste proteine si legano a catene polipeptidiche parzialmente ripiegate e le aiutano a progredire lungo la via di ripiegamento energeticamente più favorevole. Nelle condizioni affollate del citoplasma i chaperoni impediscono alle regioni idrofobiche, temporaneamente esposte nelle proteine appena sintetizzate, di associarsi fra loro per formare aggregati proteici . Tuttavia la forma tridimensionale definitiva della proteina è ancora specificata dalla sua sequenza di amminoacidi: i chaperoni semplicemente rendono il processo di ripiegamento più affidabile.
Le proteine hanno una grande varietà di forme e sono lunghe in genere da 50 a 2000 amminoacidi. Le proteine più grandi di solito consistono di parecchi domini proteici distinti (unità strutturali che si ripiegano in modo più o meno indipendente l’una dall’altra, come vedremo più avanti). La struttura dettagliata anche di un piccolo dominio è complicata e per chiarezza vengono usati convenzionalmente diversi modi per rappresentarla, ciascuno dei quali mette in evidenza aspetti differenti della proteina. Le descrizioni delle strutture proteiche sono rese più semplici dal fatto che le proteine sono costituite da combinazioni di parecchi motivi strutturali comuni, di cui ci occuperemo adesso.
L’α elica e il foglietto β sono schemi comuni di ripiegamento
Quando si confrontano le strutture tridimensionali di molte molecole proteiche diverse diventa chiaro che, sebbene la conformazione globale di ciascuna proteina sia unica, spesso si trovano in parti di esse due schemi regolari di ripiegamento. Entrambi gli schemi sono stati scoperti più di sessant’anni fa grazie a studi eseguiti sui peli e sulla seta. Il primo schema di ripiegamento a essere stato scoperto, chiamato α elica, è stato trovato nella proteina α-cheratina, che è abbondante nella pelle e nei suoi derivati, come peli, unghie e corna. Dopo meno di un anno dalla scoperta dell’α elica, una seconda struttura ripiegata, chiamata foglietto β, fu trovata nella proteina fibroina, il costituente principale della seta. Questi due schemi sono particolarmente comuni perché derivano dalla formazione di legami idrogeno fra i gruppi NOH e CPO dell’ossatura polipeptidica, senza coinvolgere le catene laterali degli amminoacidi.
Quindi, sebbene incompatibili con alcune catene laterali di amminoacidi, questi schemi possono essere formati da molte sequenze diverse di amminoacidi. In ciascun caso la catena proteica adotta una conformazione ripetitiva regolare. Il nucleo di molte proteine contiene estese regioni a foglietto β. Questi foglietti β si possono formare da catene polipeptidiche adiacenti che corrono nella stessa direzione (catene parallele) o da una catena polipeptidica che si ripiega avanti e indietro su se stessa, con ciascuna sezione della catena che corre nella direzione opposta a quella della catena più vicina (catene antiparallele).
Entrambi i tipi di foglietto β producono una struttura molto rigida, tenuta insieme da legami idrogeno che connettono i legami peptidici di catene adiacenti. Un’α elica si genera quando una singola catena polipeptidica si avvolge su se stessa per formare un cilindro rigido. Si forma un legame idrogeno fra un legame peptidico e il quarto successivo, collegando il CPO di un legame peptidico con l’NOH di un altro. Ciò dà origine a un’elica regolare con un giro completo ogni 3,6 amminoacidi.
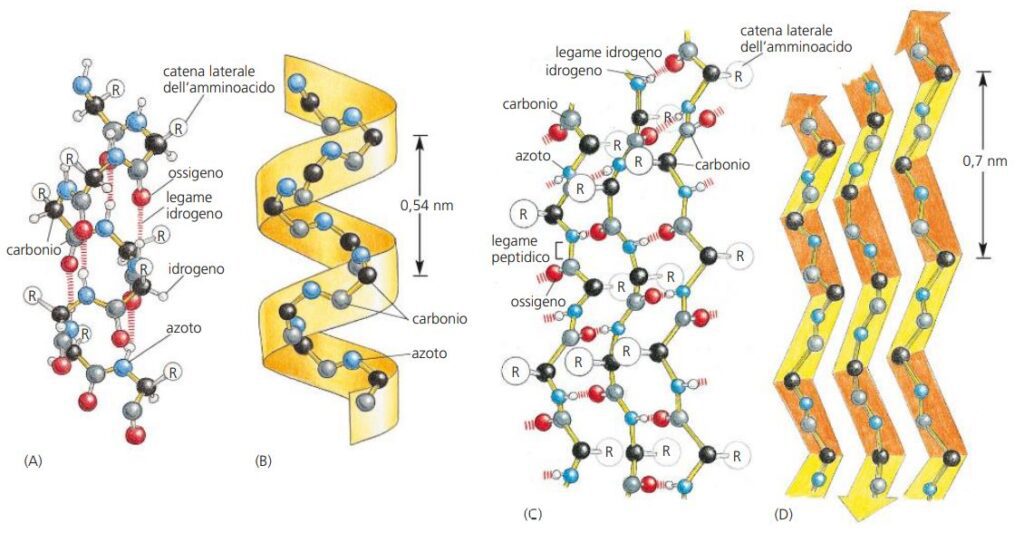
Regioni ad α elica sono abbondanti in proteine poste nelle membrane cellulari, come proteine di trasporto e recettori. Quelle porzioni di una proteina transmembrana che attraversano il doppio strato lipidico in genere lo attraversano sotto forma di un’α elica composta in gran parte da amminoacidi con catene laterali non polari. L’ossatura polipeptidica, che è idrofilica, forma legami idrogeno con se stessa nell’α elica ed è schermata dall’ambiente lipidico idrofobico della membrana dalle catene laterali non polari. In altre proteine le a eliche si avvolgono l’una intorno all’altra per formare una struttura particolarmente stabile, nota come coiled coil.
Questa struttura si può formare quando le due (o in qualche caso tre o quattro) a eliche hanno la maggior parte delle loro catene laterali non polari (idrofobiche) su un lato, così che possono avvolgersi l’una intorno all’altra, con queste catene laterali rivolte all’interno. Lunghi coiled coil a bastoncino forniscono la base strutturale di molte proteine allungate. Esempi di ciò sono l’α-cheratina, che forma le fibre intracellulari che rinforzano lo strato esterno della pelle e delle sue appendici, e la miosina, responsabile della contrazione muscolare.
I domini proteici sono unità modulari che costituiscono le proteine più grandi
Anche una piccola molecola proteica è costituita da migliaia di atomi uniti insieme da legami covalenti e non covalenti orientati precisamente. I biologi riescono a visualizzare queste strutture così complicate mediante diversi sistemi grafici tridimensionali computerizzati. Gli scienziati distinguono quattro livelli di organizzazione nella struttura di una proteina. La sequenza degli amminoacidi è nota come struttura primaria della proteina. Tratti di catena polipeptidica che formano α eliche e foglietti β costituiscono la struttura secondaria della proteina. L’organizzazione tridimensionale completa di una catena polipeptidica viene talvolta chiamata struttura terziaria e, se una particolare proteina è composta da un complesso di più di una catena polipeptidica, la struttura completa è definita struttura quaternaria.
Lo studio della conformazione, della funzione e dell’evoluzione delle proteine ha anche rivelato l’importanza centrale di un’unità di organizzazione distinta dalle quattro appena descritte. Questa è il dominio proteico, una sottostruttura prodotta da qualunque parte di una catena polipeptidica che si possa ripiegare indipendentemente in una struttura compatta stabile. Un dominio in genere contiene dai 40 ai 350 amminoacidi ed è l’unità modulare da cui sono costituite molte proteine più grandi. I diversi domini di una proteina spesso sono associati a differenti funzioni. Le molecole proteiche più piccole contengono soltanto un singolo dominio, mentre le proteine più grandi possono contenere anche parecchie decine di domini, in genere connessi fra loro da brevi tratti relativamente non strutturati di catena polipeptidica che può agire come una cerniera flessibile tra i domini.

da domini multipli. Nella proteina
Src qui rappresentata un dominio
C-terminale con due lobi (giallo e
arancione) forma un enzima proteina
chinasi, mentre i domini SH2 e
SH3 svolgono funzioni regolatrici.
(A) Un modello a nastro, con l’ATP
substrato in rosso. (B) Un modello
a spazio pieno, con l’ATP substrato
in rosso. Si noti che il sito che lega
l’ATP è posizionato all’interfaccia dei
due lobi che formano la chinasi.
Poche delle molte catene polipeptidiche possibili sono utili per le cellule
Poiché ciascuno dei 20 amminoacidi è chimicamente distinto e ciascuno può, in linea di principio, trovarsi in qualunque posizione di una catena proteica, vi sono 20 x 20 x 20 x 20 = 160000 catene polipeptidiche possibili lunghe quattro amminoacidi, o 20n catene polipeptidiche possibili lunghe n amminoacidi. Per una lunghezza tipica di una proteina di circa 300 amminoacidi si potrebbero teoricamente costruire più di 10390 (20n) diverse catene polipeptidiche. Questo è un numero talmente enorme che produrre anche una sola molecola di ciascun tipo richiederebbe molti più atomi di quanti ne esistano nell’universo. Soltanto una frazione piccolissima di questa enorme serie di catene polipeptidiche concepibili adotterebbe una conformazione tridimensionale singola stabile (secondo alcune stime, meno di una su un miliardo).
Eppure, la maggior parte delle proteine presenti nelle cellule adotta conformazioni uniche e stabili. Com’è possibile? La risposta si trova nella selezione naturale. È improbabile che una proteina con una struttura e un’attività biochimica variabile in modo non prevedibile aiuti la sopravvivenza della cellula che la contiene. Queste proteine sarebbero quindi state eliminate dalla selezione naturale nel lunghissimo processo di prova ed errore che è alla base dell’evoluzione biologica. Poiché l’evoluzione ha selezionato la funzione delle proteine negli organismi viventi, la sequenza degli amminoacidi di una proteina odierna è tale da produrre una singola conformazione estremamente stabile. Inoltre questa conformazione ha proprietà chimiche finemente regolate per permettere alla proteina di svolgere una particolare funzione catalitica o strutturale nella cellula.
Le proteine sono costruite in modo così preciso che il cambiamento anche di pochi atomi in un amminoacido può talvolta alterare la struttura dell’intera molecola in modo così grave da farle perdere completamente la funzione e quando accadono certi rari eventi di ripiegamento errato della proteina, le conseguenze possono essere disastrose per l’organismo in cui essi si verificano.
Le proteine possono essere classificate in molte famiglie
Una volta che una proteina che si ripiega in una conformazione stabile con proprietà utili si è evoluta, la sua struttura può essere modificata nel corso dell’evoluzione per permetterle di svolgere nuove funzioni. Questo processo è stato enormemente accelerato da meccanismi genetici che occasionalmente producono copie duplicate di geni, permettendo a una copia del gene di evolvere in modo indipendente per svolgere una nuova funzione. Questo tipo di evento si è verificato molto spesso nel passato; come risultato, molte proteine odierne possono essere raggruppate in famiglie proteiche, i cui membri hanno una sequenza di amminoacidi e una conformazione tridimensionale che assomiglia a quella degli altri membri della famiglia.
Consideriamo, per esempio, le serina proteasi, una grande famiglia di enzimi che tagliano proteine (proteolitici) che comprende gli enzimi digestivi chimotripsina, tripsina ed elastasi, e parecchie proteasi coinvolte nella coagulazione del sangue. Quando si confrontano le porzioni proteasiche di due di questi enzimi, si trova che parti delle loro sequenze di amminoacidi corrispondono. La somiglianza delle loro conformazioni tridimensionali è ancora più sorprendente: la maggior parte degli avvolgimenti e dei giri dettagliati delle loro catene polipeptidiche, che sono lunghe parecchie centinaia di amminoacidi, è praticamente identica. Le molte serina proteasi diverse hanno nonostante ciò attività enzimatiche distinte, e ciascuna taglia proteine diverse o legami peptidici fra tipi diversi di amminoacidi. Ciascuna perciò svolge una funzione differente in un organismo.

Quanto detto per le serina proteasi potrebbe essere ripetuto per centinaia di altre famiglie proteiche. In generale, la struttura dei diversi membri di una famiglia si è conservata di più della sequenza degli amminoacidi. In molti casi le sequenze degli amminoacidi si sono talmente diversificate che non si può essere sicuri della relazione familiare fra due proteine senza determinare le loro strutture tridimensionali. La proteina a2 del lievito e la proteina engrailed di Drosophila, per esempio, sono entrambe proteine regolatrici della famiglia a omeodominio. Poiché sono identiche soltanto in 17 dei 60 residui amminoacidici dei loro omeodomini, la loro relazione è diventata certa soltanto quando sono state confrontate le loro strutture tridimensionali. Molti esempi simili mostrano che due proteine con più del 25% di identità nella loro sequenza amminoacidica di solito hanno la stessa struttura generale.
I vari membri di una grande famiglia proteica hanno spesso funzioni distinte. Alcuni dei cambiamenti di amminoacidi che rendono diversi i membri della famiglia sono stati senza dubbio selezionati nel corso dell’evoluzione perché hanno portato a modificazioni utili nell’attività biologica, conferendo ai singoli membri della famiglia le diverse proprietà funzionali che hanno oggi. Ma molti altri cambiamenti di amminoacidi sono effettivamente “neutri”, non avendo né un effetto benefico né uno dannoso sulla struttura e sulla funzione base della proteina. Inoltre, poiché la mutazione è un processo casuale, devono esserci stati anche molti cambiamenti deleteri che hanno alterato la struttura tridimensionale di queste proteine in modo sufficiente da danneggiarle. Queste proteine difettose sarebbero state perdute tutte le volte che i singoli organismi che le producevano avevano uno svantaggio tale da essere eliminati dalla selezione naturale.
Le famiglie proteiche si riconoscono facilmente quando si sequenzia il genoma di un organismo; per esempio, la determinazione della sequenza del DNA dell’intero genoma umano ha rivelato che noi possediamo circa 21 000 geni che codificano proteine. (Si noti tuttavia che, come risultato dello splicing alternativo, le cellule umane possono produrre più di 21 000 proteine diverse). Tramite confronti di sequenze, si possono assegnare i prodotti di circa il 40% di questi geni a strutture proteiche note, appartenenti a più di 500 famiglie proteiche diverse.
La maggior parte delle proteine di ciascuna famiglia si è evoluta per svolgere funzioni un po’ differenti, come per gli enzimi elastasi e chimotripsina. Questi geni sono talvolta chiamati paraloghi per distinguerli dalle proteine corrispondenti in organismi diversi (ortologhi, come l’elastasi umana e di topo). Grazie alle tecniche potenti di cristallografia ai raggi X e di risonanza magnetica nucleare (NMR) oggi conosciamo la forma tridimensionale, o conformazione, di più di 100 000 proteine. Confrontando attentamente le conformazioni di queste proteine, i biologi strutturali (cioè gli esperti della struttura delle molecole biologiche) hanno concluso che esiste un numero limitato di modi in cui i domini proteici si ripiegano in natura, forse meno di 2000, se consideriamo tutti gli organismi.
Per la maggior parte di queste strutture, chiamate ripiegamenti proteici, è stato possibile determinare le strutture rappresentative. Il database attuale di sequenze proteiche note contiene più di venti milioni di voci e sta crescendo molto rapidamente man mano che vengono sequenziati altri genomi, rivelando un enorme numero di nuovi geni che codificano proteine. L’intervallo di dimensione dei polipeptidi codificati è molto ampio, da polipeptidi di 6 amminoacidi a proteine gigantesche di 33 000 amminoacidi. Confronti fra proteine sono importanti perché strutture correlate spesso implicano funzioni correlate. Si possono risparmiare anni di esperimenti scoprendo che una nuova proteina ha un’omologia nella sequenza amminoacidica con una proteina di cui è nota la funzione. Queste relazioni di sequenza, per esempio, hanno indicato per la prima volta che certi geni che provocano la trasformazione cancerosa delle cellule dei mammiferi codificano proteina chinasi.
Alcuni domini proteici formano parti di molte proteine diverse
la maggior parte delle proteine è composta da una serie di domini proteici, in cui regioni diverse della catena polipeptidica si sono ripiegate in modo indipendente formando strutture compatte. Si pensa che queste proteine multidominio si siano originate quando le sequenze di DNA che codificano ciascun dominio si sono unite accidentalmente, creando un nuovo gene. Molte grandi proteine si sono evolute per unione di domini preesistenti in nuove combinazioni, un processo evolutivo chiamato rimescolamento dei domini. Nuove superfici di legame si sono spesso create in corrispondenza della giustapposizione dei domini e molti dei siti funzionali in cui le proteine legano piccole molecole si trovano in quei punti.
Una sottoserie di domini proteici è stata particolarmente mobile durante l’evoluzione; sembra che questi domini abbiano strutture molto versatili e sono talvolta chiamati moduli proteici. Un aspetto importante dei domini proteici che ne spiega l’utilità è la facilità con cui possono essere integrati in altre proteine. Quando il DNA che codifica un dominio di questo tipo subisce una duplicazione in tandem, che non è insolita nell’evoluzione dei genomi, i moduli duplicati con questa disposizione “in linea” possono essere facilmente collegati in serie per formare strutture estese, sia con se stessi che con altri domini in linea. Rigide strutture estese composte da una serie di domini sono comuni specialmente nelle molecole della matrice extracellulare e nelle porzioni extracellulari di recettori proteici della superficie cellulare. Altri domini, sono di un tipo “a spina”, con gli N- e i C-terminali vicini fra loro.
Dopo riarrangiamenti genomici, questi moduli sono in genere disposti come un’inserzione in una regione ad ansa di una seconda proteina. Un confronto della frequenza relativa dell’utilizzo dei domini in eucarioti diversi ha rivelato che per molti domini comuni, come le proteina chinasi, la frequenza è simile in organismi così diversi come il lievito, i vegetali, i vermi, le mosche e gli esseri umani.
Ma ci sono eccezioni notevoli, come il dominio di riconoscimento dell’antigene del complesso maggiore di istocompatibilità (MHC) che si trova in 57 copie negli esseri umani, ma è assente negli altri quattro organismi appena menzionati. Presumibilmente questi domini hanno funzioni specializzate che non sono condivise da altri eucarioti, avendo subito una forte selezione durante l’evoluzione per poter dare origine alle copie multiple osservate. In modo simile si potrebbe supporre che un dominio come SH2, che mostra un insolito aumento di numero negli eucarioti superiori, sia particolarmente utile per la pluricellularità.
Certe coppie di domini si trovano insieme in molte proteine
Possiamo costruire una grande tavola che mostra l’utilizzo dei domini in ciascun organismo di cui conosciamo la sequenza del genoma. Per esempio, si stima che il genoma umano contenga circa 1000 domini immunoglobulinici, 500 domini di proteina chinasi, 250 omeodomini che legano il DNA, 300 domini SH3 e 120 domini SH2. Inoltre, emerge che più dei due terzi delle proteine consistono di due o più domini e che le stesse coppie di domini si trovano ripetutamente nella stessa disposizione relativa in una proteina. Sebbene metà di tutte le famiglie di domini sia comune ad archei, batteri ed eucarioti, soltanto il 5% delle combinazioni di due domini è ugualmente condiviso. Questo schema suggerisce che la maggior parte delle proteine che contengono combinazioni particolarmente utili di due domini sia comparsa relativamente tardi nel corso dell’evoluzione.
Il genoma umano codifica una serie complessa di proteine, la funzione di molte delle quali è sconosciuta
Il risultato del sequenziamento del genoma umano è stato sorprendente, perché rivela che i nostri cromosomi contengono soltanto circa 21 000 geni codificanti proteine. In base al numero dei geni sembriamo non essere più complessi della minuscola erba Arabidopsis e soltanto circa 1,3 volte più complessi di un nematode. Le sequenze del genoma rivelano anche che i vertebrati hanno ereditato quasi tutti i loro domini proteici dagli invertebrati; soltanto il 7% dei domini umani identificati è specifico dei vertebrati. Tuttavia ciascuna proteina umana è in media più complicata. Un processo di rimescolamento di domini durante l’evoluzione dei vertebrati ha dato origine a molte nuove combinazioni di domini proteici, con il risultato che nelle proteine umane vi è quasi il doppio delle combinazioni di domini che si trovano in un verme o in una mosca.
Così, per esempio, il dominio serina proteasi simile a tripsina è collegato ad almeno altri 18 tipi di domini proteici nelle proteine umane, mentre si trova unito covalentemente a 5 soli domini diversi nel verme. Questa varietà extra nelle nostre proteine aumenta di molto la gamma di possibili interazioni proteina-proteina, ma come contribuisca a renderci umani non è noto. La complessità degli organismi viventi è impressionante e sapere che al momento non abbiamo il minimo indizio su quale potrebbe essere la funzione di più di 10 000 delle proteine che abbiamo identificato finora nel genoma umano smorza alquanto l’entusiasmo. La prossima generazione di biologi cellulari dovrà affrontare certamente enormi sfide e non mancheranno affascinanti misteri da risolvere.
Le molecole proteiche più grandi spesso contengono più di una catena polipeptidica
Gli stessi deboli legami non covalenti che rendono una catena proteica capace di ripiegarsi in una conformazione specifica permettono anche alle proteine di legarsi fra loro per produrre strutture più grandi nella cellula. Qualunque regione della superficie di una proteina che può interagire con un’altra molecola attraverso serie di legami non covalenti è chiamata sito di legame. Una proteina può contenere siti di legame per varie molecole, sia grandi che piccole. Se un sito di legame riconosce la superficie di una seconda proteina, lo stretto legame di due catene polipeptidiche ripiegate in corrispondenza di questo sito crea una molecola proteica più grande con una geometria precisamente definita. Ciascuna catena polipeptidica in una proteina di questo tipo è detta subunità proteica.
Nel caso più semplice due identiche catene polipeptidiche ripiegate si legano fra loro in una disposizione “testa-testa”, formando un complesso simmetrico di due subunità proteiche (un dimero) tenuto insieme da interazioni fra due siti di legame identici. La proteina repressore Cro – una proteina virale regolatrice dei geni che si lega al DNA per spegnere geni virali in una cellula batterica infettata – fornisce un esempio. Nelle cellule si trovano comunemente molti altri tipi di complessi proteici simmetrici, formati da copie multiple di una singola catena polipeptidica. Molte proteine cellulari contengono due o più tipi di catene polipeptidiche. L’emoglobina, la proteina che trasporta ossigeno nei globuli rossi, contiene due subunità identiche di a-globina e due subunità identiche di b-globina, disposte simmetricamente. Queste proteine con molte subunità sono molto comuni nelle cellule e possono essere molto grandi.
Fonti
- Anfinsen CB (1973) Principles that govern the folding of protein chains. Science 181, 223-230.
- Caspar DLD e Klug A (1962) Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 27, 1-24.
- Dill KA e MacCallum JL (2012) The protein-folding problem, 50 years on. Science 338, 1042-1046.
- Eisenberg D (2003) The discovery of the alpha-helix and betasheet, the principal structural features of proteins. Proc. Natl Acad. Sci. USA 100, 11207-11210.
- Fraenkel-Conrat H e Williams RC (1955) Reconstitution of active tobacco mosaic virus from its inactive protein and nucleic acid components. Proc. Natl Acad. Sci. USA 41, 690-698.
- Goodsell DS e Olson AJ (2000) Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 29, 105-153.
- Greenwald J e Riek R (2010) Biology of amyloid: structure, function, and regulation. Structure 18, 1244-1260.
- Harrison SC (1992) Viruses. Curr. Opin. Struct. Biol. 2, 293-299.
- Hudder A, Nathanson L e Deutscher MP (2003) Organization of mammalian cytoplasm. Mol. Cell. Biol. 23, 9318-9326.
- Kato M, Han TW, Xie S et al. (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149, 753-767.
- Koga N, Tatsumi-Koga R, Liu G et al. (2012) Principles for designing ideal protein structures. Nature 491, 222-227.
- Li P, Banjade S, Cheng H-C et al. (2012) Phase transitions in the assembly of multivalent signalling proteins. Nature 483, 336- 340.
- Lindquist SL e Kelly JW (2011) Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases – progress and prognosis. Cold Spring Harb. Perspect. Biol. 3, a004507.
- Maji SK, Perrin MH, Sawaya MR et al. (2009) Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 325, 328-332.
- Nelson R, Sawaya MR, Balbirnie M et al. (2005) Structure of the cross-β spine of amyloid-like fibrils. Nature 435, 773-778.
- Nomura M (1973) Assembly of bacterial ribosomes. Science 179, 864-873.
- Oldfield CJ e Dunker AK (2014) Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 83, 553-584.
- Orengo CA e Thornton JM (2005) Protein families and their evolution – a structural perspective. Annu. Rev. Biochem. 74, 867-900.
- Pauling L e Corey RB (1951) Configurations of polypeptide chains with favored orientations around single bonds: two new pleated sheets. Proc. Natl Acad. Sci. USA 37, 729-740.
- Pauling L, Corey RB e Branson HR (1951) The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl Acad. Sci. USA 37, 205-211.
- Prusiner SB (1998) Prions. Proc. Natl Acad. Sci. USA 95, 13363- 13383.
- Toyama BH e Weissman JS (2011) Amyloid structure: conformational diversity and consequences. Annu. Rev. Biochem. 80, 557-585.
- Zhang C e Kim SH (2003) Overview of structural genomics: from structure to function. Curr. Opin. Chem. Biol. 7, 28-32.